

The Environmental Risk Assessment
Environmental protection has become clearly visible in society in recent years. Pharmaceutical industry has been aware of this issues for a long time. Regulatory Bodies not only focus on the patient safety, but also consider the impact of medicinal product on the whole environment. The Environmental Risk Assessment (ERA) aims to establish the potential environmental hazards arising from the use, storage and disposal of medicinal products.
When are you obligated to submit Environmental Risk Assessment (ERA)?
In accordance with Article 8(3) of Directive 2001/83/EC, every application for medicinal product for marketing authorisation in EU, through centralised, mutual recognition (MRP), decentralised (DCP) or national procedure needs to be submitted with relevant ERA authorised by an Expert. According to NTA, Vol. 2B-CTD, ERA should be presented at 1.6.1 module (for medicinal products which do not contain GMOs (Genetically Modified Organisms)), or 1.6.2 module (for products containing, or consisting of GMOs). ERA is also required for type II variations or extensions, if there is a potential impact on the environment (e.g. addition of new indication). Renewals, type IA/IB variations do not trigger the need to submit ERA unless it may result in a potential increase in active substance consumption.
SciencePharma has great experience in evaluation whether a given ERA is sufficient for EU procedures on national and international level (DCP, MRP, central). We can demonstrate what is missing, what can cause Expert questions, and finally we can comprehensively prepare the whole risk analysis. We can also advise on next steps depending on the outcome of ERA.
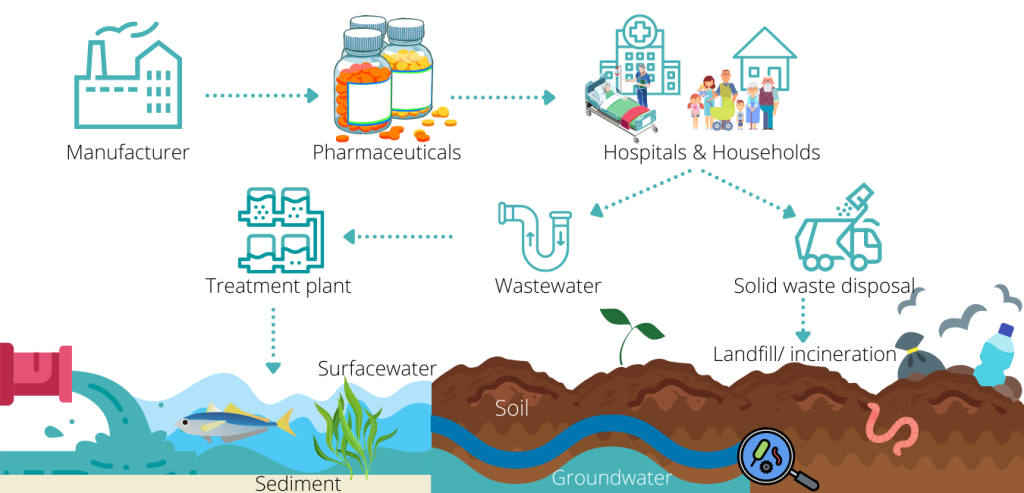
Pict. Entry paths into environment for medicinal products arising from their use, storage and disposal
What documents govern the ERA requirements?
Environmental Risk Assessment is regulated by the Guideline on the environmental risk assessment of medicinal products for human use and the Guideline on environmental risk assessments for medicinal products consisting of, or containing, genetically modified organisms (GMOs) and other, like e.g. Q&A, accompanying documents prepared by Committee for Medicinal Products for Human Use (CHMP). In general they focus on the exposure of the drug substance to environment (e.g. soil, surface and groundwater), its fate and effects in the environment (e.g. on algae, Daphnia, fish, microbes). Assessment of the potential risks to the environment consists of two phases and includes hazard assessment for persistence, bioaccumulation, toxicity (PBT) and calculation of the Predicted Environmental Concentration (PEC) followed by further analysis. If unclear of how to interpret guidelines or confused by calculations, our CMC Experts will surely help you with explanations!
Can you avoid expensive environmental tests?
Yes, there may be circumstances under which the absence of ERA studies may be justified. This is the case when a generic, hybrid or well-established use (WEU) of medicinal product is introduced to the market. Nevertheless, an appropriate rationale should always be presented, taking into consideration the impact on the environment, especially a possible significant increase of environmental exposure to the drug substance. If you wonder how to justify it, we can assist you.
Why should you use our experience?
Our Experts have deep knowledge because we have been looking at ERA from plethora of various angles, e.g. different product types, different substances, different procedure types and countries involved. If you wonder if ERA concerns your product and want to have ERA written by one of our Experts, you’re welcome to contact us.
Would you like to know more? Need help in preparing ERA? Would you like our support to justify the lack of specific ERA studies? Check our website or contact us directly.