

Impurities & Genotoxicity
Impurities and Genotoxicity: Ensuring Drug Safety and Quality
The presence of impurities in medicinal products is a critical concern for patient safety and drug quality. Understanding the sources of impurities and their potential toxicity, including genotoxicity, is essential for effective control and regulatory compliance. At SciencePharma, we offer expert services in impurities and genotoxicity assessment, helping you navigate the complex regulatory landscape and ensure the safety and efficacy of your products.
The concept of impurities and their sources
Impurities are foreign substances whose presence in a medicinal product or a raw material used to produce such a product is undesirable and should be strictly controlled. Impurities in active substances (API – Active Pharmaceutical Ingredient) and finished products may affect their quality, safety and effectiveness.
The main sources of impurities include:
- Starting materials
- Active substances and excipients
- Conditions of the production process
- Manufacturing technology
- Packaging materials
- Transport and storage conditions
- Human factor (contamination).
To avoid the negative impact of impurities on patient health, it is essential to ensure proper control of impurities in medicinal products. This is achieved through guidelines developed by the International Council for Harmonisation (ICH), including ICH Q3A and Q3B, which address the assessment and control of impurities in active substances and medicinal products.
What are the main assumptions of the ICH Q3A and Q3B guidelines?
The ICH Q3A guideline addresses the identification, classification, and establishment of limits for impurities in active substances. It distinguishes three types of impurities:
- Organic impurities – Primarily originating from chemical synthesis processes. These may include by-products of reactions, residues of starting materials, or decomposition products of the active substance.
- Inorganic impurities – Residues of metal catalysts, water, or substances originating from reagents or production equipment. (The topic of metal impurities is extensively discussed on the SciencePharma website: “Risk assessment and control of elemental impurities” as well as in the ICH Q3D guideline.)
- Solvents – Residues of organic solvents used during synthesis (the rules for the classification of solvents and the establishment of their limits are detailed in the ICH Q3C guideline).
The ICH Q3B guideline addresses the issue of impurities in finished medicinal products. These types of impurities can be divided into:
- impurities originating from API – these may be inorganic, organic substances or solvent residues not removed during the manufacturing process of the final product;
- impurities resulting from manufacturing processes – these include degradation products generated during manufacturing, storage, as a result of interaction with packaging or technological processes.
Impurity limits

The limits of impurities in both API and final products depend on the maximum daily dose (MDD) of the drug. Impurities above specific thresholds must be identified and assessed for their toxicity. Manufacturers are obliged to monitor impurity levels and conduct stability studies to determine whether these levels increase over time or under unfavourable storage conditions.
The application of the above guidelines is well understood by the experts at SciencePharma, who have prepared numerous reports so far on establishing or justifying impurity limits for their clients, which have been accepted by competent authorities. At SciencePharma, we understand the importance of patient safety, as well as the need to maintain appropriate quality and efficacy of the drug. If you’re facing similar challenges, contact us!
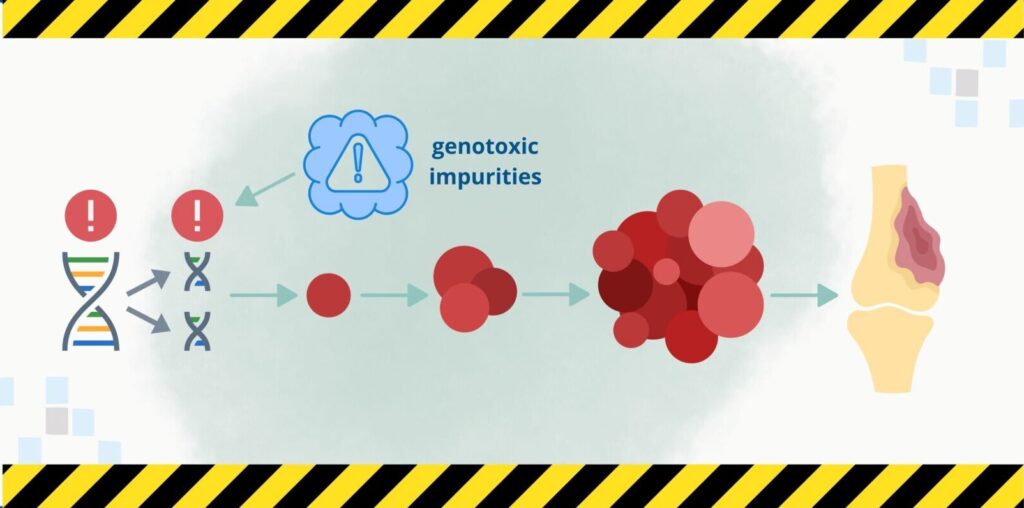
What are genotoxic impurities?
A specific type of impurities that falls outside the previously mentioned guidelines is genotoxic impurities. They pose a significant health risk to patients, as they can cause DNA mutations, potentially leading to cancer. To minimize this risk, the control of impurities with potential genotoxic effects in medicinal products has been introduced, as described in the ICH M7 guideline, which applies to both active substances and finished products.
Mutagenicity risk assessment
The ICH M7 guideline classifies genotoxic impurities based on their mutagenic potential derived from experimental data and chemical structure assessment. It distinguishes five classes:
- Class 1: Impurities with known mutagenicity and carcinogenicity
- Class 2: Impurities with mutagenic potential for which exposure limits are established
- Class 3: Potentially genotoxic substances based on their structure, requiring further analysis
- Class 4: Potentially genotoxic substances, API derivatives
- Class 5: Impurities that should be considered non-mutagenic based on available data
Methods for testing potentially mutagenic substances include both structural assessment (QSAR modelling) and biological tests (e.g. on bacterial strains – Ames test). In addition, appropriate strategies should be implemented for controlling production processes or modifying synthesis pathways to minimize the risk of these impurities.
The ICH M7 guideline introduces the concept of the threshold of toxicological concern (TTC). This threshold is expressed as an acceptable daily limit that poses a negligible (less than 1 in 100 000 cases) risk of cancer and toxic effects. It depends on the duration of therapy and the dose of the drug taken. According to the guideline, the TTC value should not exceed 1.5 µg per day.
But what if this value is exceeded? What is the procedure if the discussed impurity shows structural alerts but has no confirmed mutagenicity?
SciencePharma experts can answer these and other questions! They can help you classify impurities, set appropriate limits, or justify cases where genotoxic impurity control is unnecessary or where higher acceptance thresholds are allowed.
Why is it worth working with us?
If you need expert advice, do not hesitate to contact us!
Our team will help you:
- analyze possible sources of impurities, their potential toxicity and control methods;
- establish impurity limits, justify exceeding guideline values or determine when impurity level tests during product storage can be omitted.
SciencePharma provides comprehensive support in this area, based on extensive experience and individualized approach to each case. Our expertise enables us to reduce unnecessary analyses, saving you time and costs while maintaining high quality and compliance with regulatory requirements.
Sources:
ICH Q3A Impurities in New Drug Substances
https://www.ema.europa.eu/en/ich-q3a-r2-impurities-new-drug-substances-scientific-guideline
ICH Q3B Impurities in New Drug Products
https://www.ema.europa.eu/en/ich-q3b-r2-impurities-new-drug-products-scientific-guideline
ICH M7 Guideline on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk